La fenilcetonuria hace parte del grupo de enfermedades a tamizar en Colombia según la ley 1980. Aunque en nuestro país la incidencia de fenilcetonuria es baja, 1,33 por cada 10000 nacidos vivos, es una patología que tiene graves consecuencias en la salud de la población infantil, ya que causa retraso mental y retraso en el crecimiento.

La fenilcetonuria es una enfermedad hereditaria de carácter autosómico recesivo, en donde hay acumulación del aminoácido fenilalanina por defectos en la enzima fenilalanina hidroxilasa que lo sintetiza para convertirlo en Tirosina. Causando un daño cerebral que puede ser significativo o muy grave.
Existen dos tipos de fenilcetonuria:
1. Fenilcetonuria clásica: Es la forma más grave de la enfermedad ya que no existe la enzima que transforma la fenilalanina a tirosina o se encuentra en mínima cantidad. Causando un daño cerebral grave y una discapacidad intelectual permanente, además de convulsiones, retraso en el desarrollo, problemas de conducta y trastornos psiquiátricos permanentes.

Figura 5. Dos hermanos afectos de PKU. El hermano mayor, de 11 años, fue tratado tardíamente y está afecto de un retraso mental severo, mientras que su hermana de 2 años y medio fue tratada desde los primeros meses de vida y presenta un desarrollo neurológico normal.
2. Hiperfenilalaninemia: Es la forma leve o moderada, en donde hay una cantidad de enzima que no permite una acumulación tan alta de fenilalanina en el cerebro. Hay menor daño cerebral.

El diagnóstico de fenilcetonuria se realiza en dos etapas:
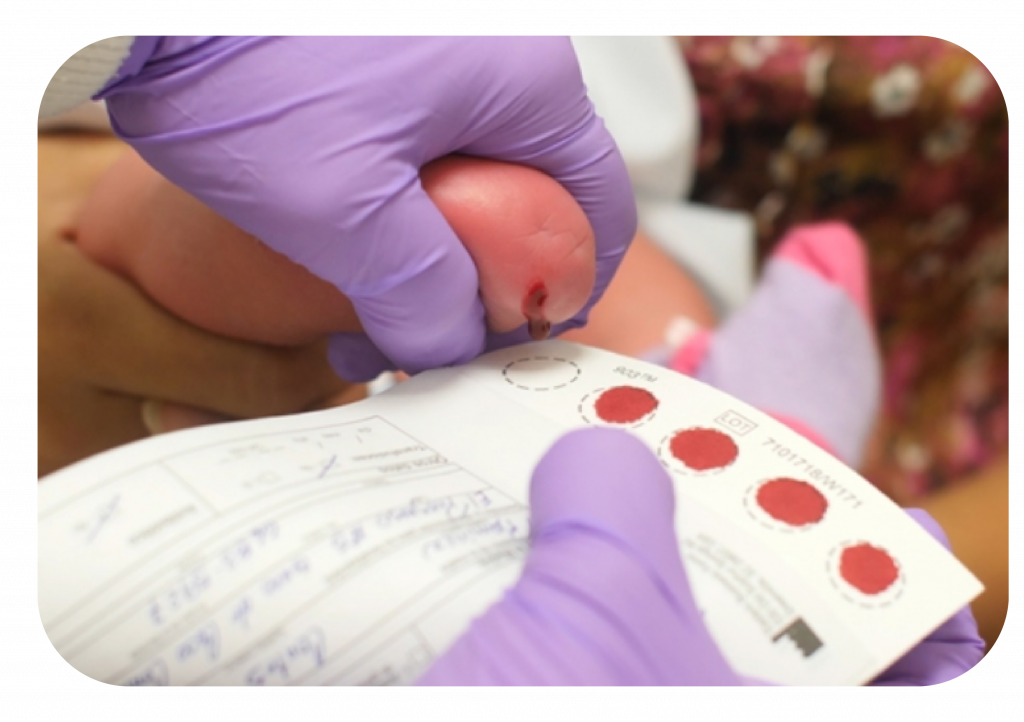
1. A través del Tamizaje Neonatal en sangre de talón se identifican los posibles casos de fenilcetonuria en los primeros días de vida. En Colombia el punto de corte para la medición de Fenilalanina es >2mg/ml.
2. Existen diversas pruebas confirmatorias para el diagnóstico de fenilcetonuria, se pueden analizar muestras de sangre de talón o sangre total con EDTA para la medición de enzimas involucradas en el proceso de transformación de fenilalanina, algunas pruebas son: Determinación de razón Fenilalanina/Tirosina, actividad enzimática 6 piruvoil tetrahidropterina sintasa, determinación de pterinas, fenilalanina en plasma, entre otras.

Perfil de aminoácidos de una muestra de sangre seca en papel filtro: PKU
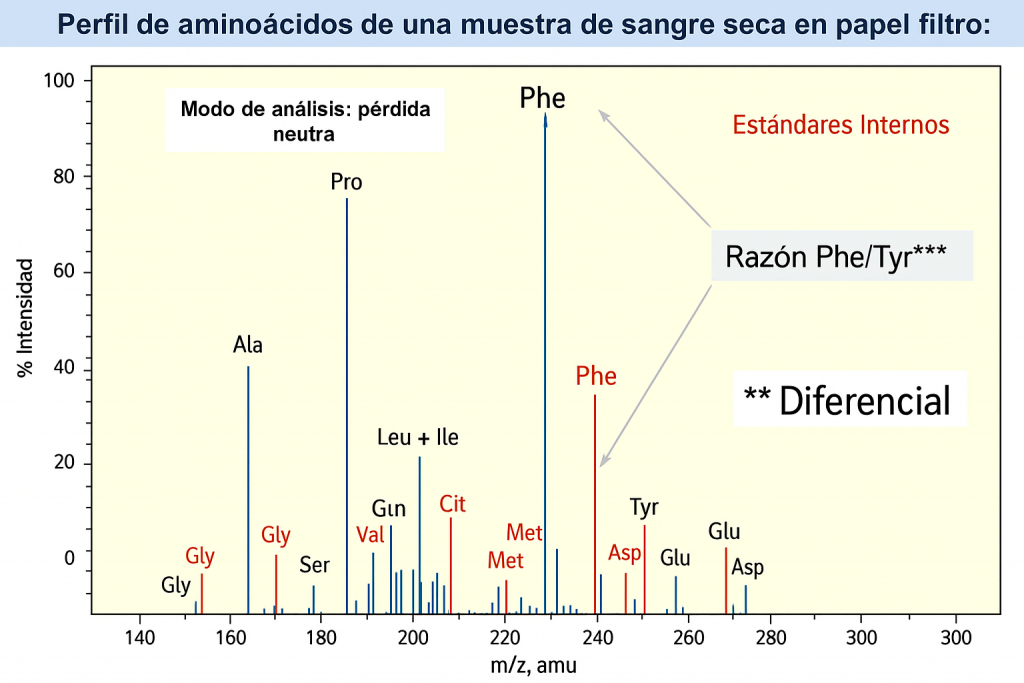
Hay grandes cantidades de fenilalanina en diversos alimentos proteínicos como la leche, los huevos y otros alimentos comunes, por lo tanto, el tratamiento es dado por nutricionista y consiste principalmente en una dieta muy limitada en proteínas; tomar leche maternizada para la fenilcetonuria (un suplemento nutricional especial), incremento del consumo de frutas y verduras y el consumo de suplementos nutricionales especiales por toda la vida del paciente.

Pirámide alimentaria en la PKU

El pronóstico de los recién nacidos que son diagnosticados a tiempo con fenilcetonuria es muy alentador orientado a una excelente
calidad de vida, ya que ppueden alcanzar un desarrollo intelectual normal y un crecimiento y desarrollo adecuado.
El tamizaje neonatal es la clave para que nuestros niños colombianos tengan una infancia feliz y un crecimiento y desarrollo adecuado.

Bibliografía
https://rarediseases.info.nih.gov/espanol/13560/fenilcetonuria
https://www.mayoclinic.org/es/diseases-conditions/phenylketonuria/diagnosis-treatment/drc-20376308
https://www.sciencedirect.com/science/article/abs/pii/S1096719209002996
https://neuropediatra.org/2016/04/25/pku-enfermedad-rara-fenilcetonuria/
https://metabolicas.sjdhospitalbarcelona.org/noticia/manual-dietetico-pku-acompanar-guia-clinica
https://prensa.css.gob.pa/2021/10/20/tamizaje-neonatal-oportunidad-de-mejor-vida-para-el-bebe/